BUILDING ARCHITECTURAL MODELS OF MULTI-PROTEIN COMPLEXES BASED ON ION MOBILITY-MASS SPECTROMETRY DATASETS.
Structural biology is ultimately concerned with determining high-resolution structures of all the functional macromolecules within living cells and tissues. While structural information can be obtained using a wide array of technologies, conventional methods face significant challenges when applied to dynamic, heterogeneous multiprotein targets. Consequently, there is a need to investigate new approaches that define the subunit stoichiometry, composition, and shape of heterogeneous macromolecular complexes of biological importance. With the advent of nano-electrospray ionization (nESI) and consequently the ability to retain native-like protein complex structures in the gas-phase [see publications], mass spectrometry (MS) is quickly becoming an essential tool within the structural biology community. In the Ruotolo group, we are developing Ion Mobility-Mass Spectrometry (IM-MS) based techniques to answer important structural questions about these large multiprotein machines. What sets our technology apart from traditional mass spectrometry is our unique ability to measure the mass, charge, and collision cross-section of an ion simultaneously. With the added dimension of cross section in our data sets, we are able to carry out detailed structural studies on a wide range of protein complexes in a high throughput manner. Our efforts focused in structural characterization of protein complexes fall into three main categories: understanding the structural properties of proteins in the gas-phase, using IM-MS measurements to develop structural models of protein complexes, and developing computational platforms for integrating IM-MS and other datasets into 3D models.
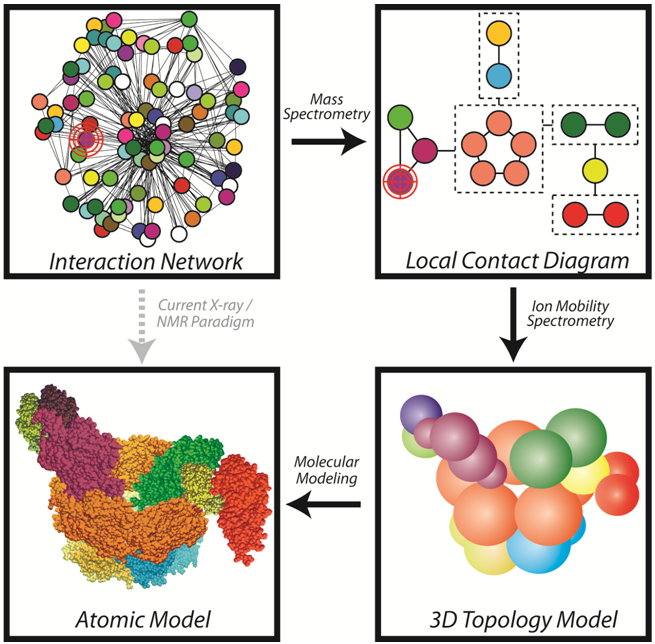
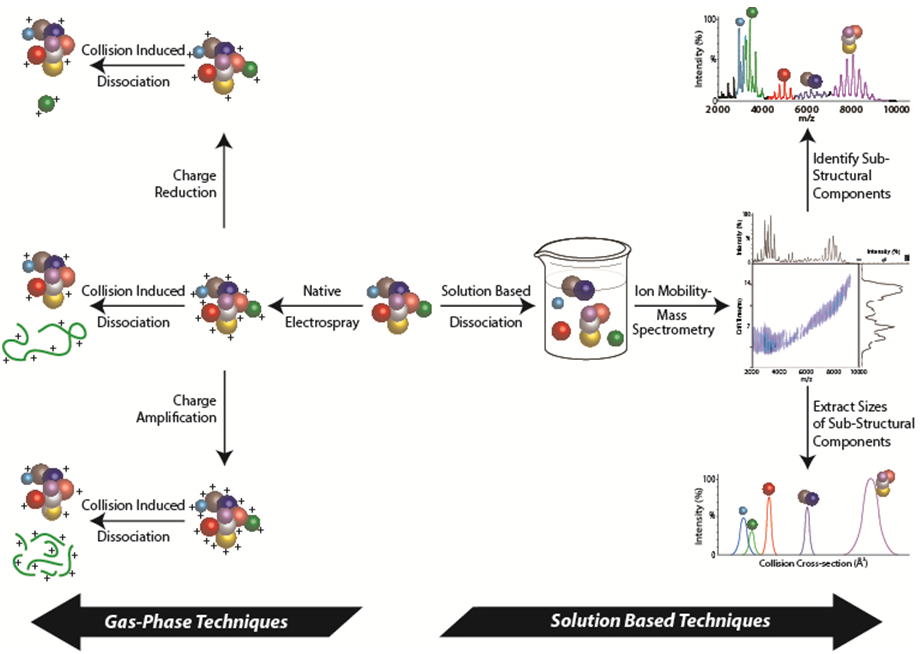
The recent explosion in proteomics data has left the community with detailed interaction maps and an overwhelming amount of new structural biology targets. Using IM-MS techniques that define the connectivity and stoichiometry of a complex through the generation of sub-complexes (see Figure), our group has developed robust methods for generating topology models of large protein complexes (see publication list). Using nano-electrospray ionization (nESI), protein complexes introduced into the gas-phase can retain their native folds and 3D arrangement under optimized conditions. This allows for the accurate determination of the protein complex stoichiometry based on detailed mass measurements (see Group Instrumentation). By manipulating protein complexes both in solution (prior to nESI) and in the gas-phase, protein interfaces can disrupted to produce many smaller subcomplexes. Iterative measurements of subcomplex stoichiometry and composition allow us to generate 2D contact diagrams for unknown protein complexes. Complexes can also be subjected to collision induced dissociation (CID) and charge manipulation strategies to gain further connectivity information. Furthermore, by using highly accurate ion mobility measurements, the orientiationally-averaged sizes of both intact and dissociated complexes can be used to produce distance and angle restraints for each subunit within a given assembly.
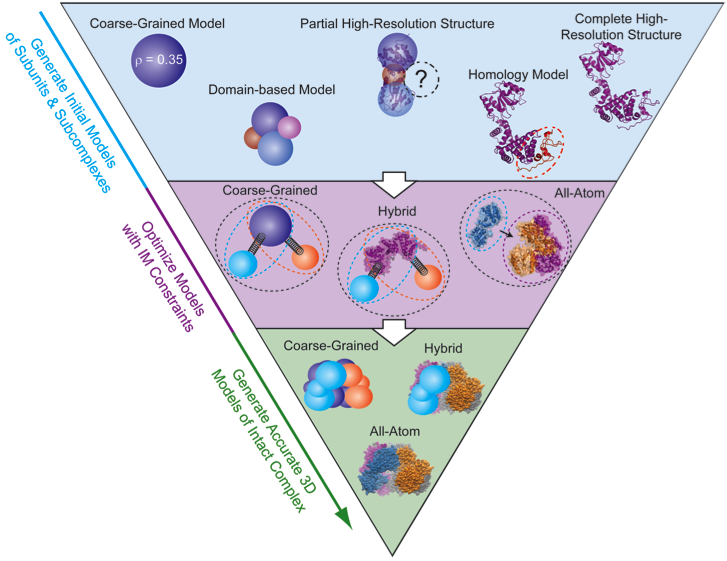
By combining the data generated by IM-MS with bioinformatics, molecular mechanics, and molecular dynamics we are positioned to generate a full, low-resolution 3D architecture for protein complexes. 3D modeling begins by bringing together all known structural information for the constituent proteins. First, each protein must have a representation in the model. When a subunit structure is unknown, it can be represented as a sphere with a radius corresponding to its IM-derived size value. At the other end of the spectrum, if an atomic resolution model of the protein exists it can be integrated directly into the modeling algorithm or be added to the final model later to reduce computational costs. There are also a variety of intermediate representations that can be used simultaneously including partial atomic structures, homology models, or domain-level representations. IM-MS data is encoded in the form of distance and connectivity restraints between each subunit within the assembly. In the simplest example, if the size of two individual proteins is known, as well as the size of their dimer, a center-to-center distance restraint can be applied that keeps the proteins at a specific distance characteristic of their interface. IM-MS restraints can be used to restrain distances, angles, and ultimately the shapes of intact complexes. We have developed a number of python scripts that build on previous work (see references 42 and 60) for generating and scoring models based on IM-MS inputs, and in the future, we hope to generate a user-friendly modeling platform that allows easy translation of IM-MS inputs into spatial restraints and allows for rigorous scoring of the precision of the structures generated.
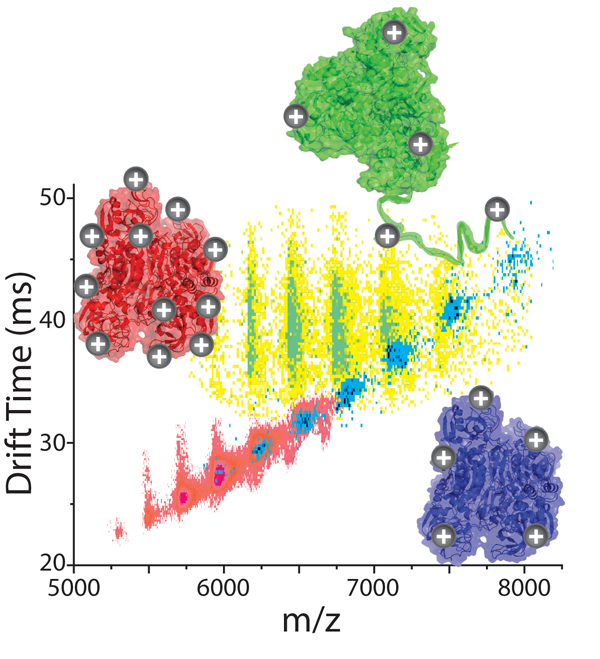 Many reports have indicated that the overall charge of the ions observed innESI-MS experiments can have a significant effect on the information content of the IM-MS dataset obtained. For example, it has been observed that the quaternary structure of protein complexes is altered from its native form in more highly-charged ions in some cases (see publications). Also, evidence from collision induced dissociation (CID) has recently revealed dramatic charge-dependant mechanisms for product ion generation (see publications). The utility associated with charge manipulation is clear and we are in the process of critically evaluating a number of charge reduction approaches adapted for IM-MS analysis of protein complexes.
Many reports have indicated that the overall charge of the ions observed innESI-MS experiments can have a significant effect on the information content of the IM-MS dataset obtained. For example, it has been observed that the quaternary structure of protein complexes is altered from its native form in more highly-charged ions in some cases (see publications). Also, evidence from collision induced dissociation (CID) has recently revealed dramatic charge-dependant mechanisms for product ion generation (see publications). The utility associated with charge manipulation is clear and we are in the process of critically evaluating a number of charge reduction approaches adapted for IM-MS analysis of protein complexes.
Our preliminary data shown here indicates that different methods of charge reduction can provide different gas-phase structures for multi-protein complexes. Here, data for alcohol dehydrogenase tetramers (144 kDa) is charge reduced using two strategies and compared to control datasets (red). In a solution-phase additive strategy (shown in green), base is added to the complexes in solution, and collisional activation in the gas-phase is required to remove the large number of base molecules adhered to the protein complex and initiate charge reduction. This activation step causes the complex to unfold. The same base molecules nebulized into the ion source and allowed to react with the complex ions in the gas-phase produces compact gas-phase structures, similar to the native structure of the assembly (shown in blue).