|

|
Effect of Axial Thiolate Coordination on the Properties of Ferrous Heme-Nitrosyls
Using electron paramagnetic resonance (EPR) spectroscopy and density functional (DFT) calculations, we explored the effect of
thiolate coordination to ferrous heme nitrosyls. NO bound heme centers with axial cysteinate coordination are present
in fungal nitric oxide reductase (P450nor) and nitric oxide synthase (NOS) and hence, are of great biological significance.
From the literature, it is known for a long time that the binding of an axial N-donor ligand has a profound effect on the electronic
structure of the Fe(II)-NO unit as evidenced by a strong change of the EPR spectrum [1].
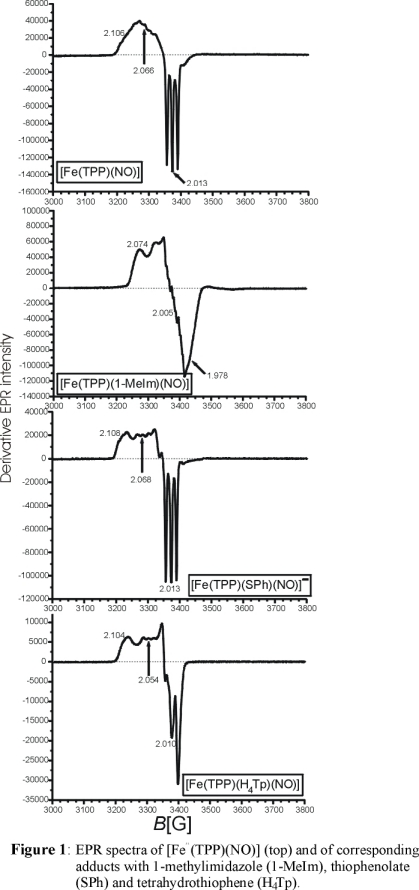
For the five-coordinate (5C) complex (Figure 1, top), a characteristic spectrum is observed with hyperfine lines of the 14N
of the coordinated NO ligand on the smallest g value g(min). Since 14N has a nuclear spin of I = 1, this leads to the
appearance of three lines in the spectrum. The six-coordinate (6C) complex with bound 1-methylimidazole shows a broader spectrum where
hyperfine lines are observed for the nitrogens of NO and of the trans-N donor on the medium g value g(mid). This can be
explained taking into account the
difference in electronic structure between the 5C and 6C complexes.
DFT calculations show that the g tensor has a different orientation in these complexes which causes the change in the
hyperfine [2],[3]. In order to explore the effect of thiolate coordination, we synthesized corresponding adducts of type
[Fe(TPP)(L)(NO)] (TPP = tetraphenylporphyrin dianion; L = thiophenolate derivatives, thioether) and measured their EPR spectra in situ.
EPR spectra obtained with thiophenolate (PhS-) and tetrahydrothiophene (H4Tp) as sixth ligands
are shown in Figure 1. These spectra mostly resemble the data obtained for the 5C complex [Fe(TPP)(NO)],
which indicates that the interaction of these ligands with Fe(II) is weak. On the other hand, EPR spectra obtained for
the ferrous heme NO adducts of P450cam and P450nor [4],[5] are comparable with those obtained for [Fe(TPP)(1-MeIm)(NO)],
which indicates that in these cases the interaction is strong. Further studies are now pursued to obtain model complexes that
better resemble the enzymatic species.
DFT calculations on the model system [Fe(P)(SMe)(NO)]
(P = porphine, MeS- is a simple model for cysteinate) for the biological species show that the trans-effect
of the thiolate on the bound NO is stronger than that of N-donor ligands like 1-methylimidazole. This further weakens the
Fe-NO bond and should therefore lead to less tightly bound NO in the presence of an axial thiolate. Importantly, these predictions
are in agreement with the experimental rates of NO loss for ferrous heme NO adducts determined for a number of
corresponding proteins [6].
We have now started to also investigate corresponding ferric heme-nitrosyls with axial thiolate coordination. Ferric heme-nitrosyls
with axial N-donor ligands like histidine in proteins or imidazole, pyridine, etc. in model complexes bound trans to NO show linear Fe-N-O units
and N-O and Fe-NO stretching frequencies around 1900-1930 cm-1 and 580-600 cm-1, respectively. We have studied the
vibrational and electronic properties of ferric heme-nitrosyls in detail.
In comparison, the presence of an axial thiolate ligand leads to interesting variations in the geometric and electronic properties of these complexes, in particular,
a distinct bending of the Fe-N-O unit with Fe-N-O angles of ~160o is observed. This was found both in the ferric NO adduct of the enzyme P450nor, but
also in the model complex [Fe(OEP)(SR-H2)(NO)] (SR-H2 = S-2,6-(CF3CONH)2C6H3) [7]. In comparison, [Fe(OEP)(N-donor)(NO)]+ complexes with the same porphyrin
exhibit linear Fe-N-O geometries, pointing directly towards the thiolate as the cause for Fe-NO bending. In addition, vibrational data show that both the N-O and the Fe-NO
stretch are shifted to distinctively lower energy in the thiolate-coordinated complexes, both in proteins and in model systems. For example, the model complex [Fe(OEP)(SR-H2)(NO)]
exhibits ν(N-O) and ν(Fe-NO) at 1850 and 549 cm-1, respectively. Importantly, a plot of the Fe-NO versus the N-O
vibrational frequencies for the known thiolate-coordinated ferric heme-nitrosyls reveals a direct correlation of these vibrational energies as shown in Figure 2.
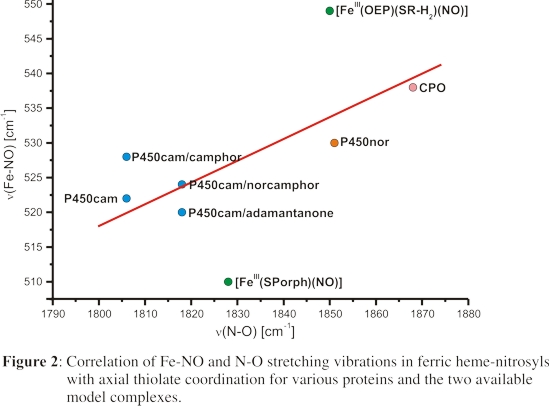
This is surprising as in the generally accepted electronic structure description of ferric heme-nitrosyls, the Fe-N-O unit is described as having Fe(II)-NO+ character
with dominant π-backbonding (see here). Hence, changes in the Fe-NO bond should
lead to an inverse correlation of the Fe-NO and N-O vibrational frequencies. The direct correlation, evident from Figure 2, implies changes in the Fe-NO σ bond
along this series of complexes, the origin of which was later identified to stem from a σ-trans effect of the strongly donating thiolate ligand.
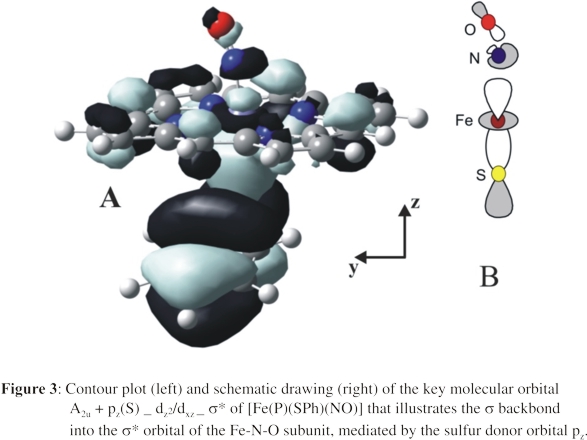
DFT calculations further support the idea that the bending of the Fe-N-O unit in the presence of an axial thiolate ligand as well as the simultaneous weakening of the Fe-NO and N-O bonds
(reflected by the corresponding vibrational stretching frequencies) are due to a trans effect (more specifically, trans interaction, since this is a thermodynamic effect) of the thiolate ligand. First, the computational results show that the ground state of these complexes
is best described as Fe(II)-NO+, just as in the case of the ferric heme-nitrosyls with axial imidazole or N-donor coordination. Hence, the Fe-NO interaction is dominated
by π-backbonding between Fe(II) and NO+. However, the observed change in the Fe-NO bond cannot simply correspond to a change in this π-backbonding
interaction, as this would lead to an inverse correlation of the Fe-NO and N-O bond strengths, and hence, vibrational stretching frequencies. This is not observed in Figure 2. This raises
the question which orbital interaction could be responsible for this curious observation. A detailed analysis of the obtained wavefunctions from the DFT calculations shows that this effect
is actually correlated to backbonding into a σ* orbital of the Fe-N-O unit as illustrated in Figure 3. This σ* orbital is fully antibonding
with respect to the Fe-N-O unit, and normally unoccupied. However, this orbital becomes partially occupied by admixture into the occupied, high-lying σ donor orbital of the
anionic trans ligand to NO. The stronger the σ-donor properties of the proximal ligand, i.e. the higher the corresponding donor orbital is in energy, the more pronounced
this admixture becomes, and the more the Fe-N-O unit bends and the weaker the Fe-NO and N-O bonds become. This unusual σ-backbond into the σ* orbital
of the Fe-N-O unit, mediated by the σ-donor orbital of the anionic trans ligand to NO, is a new orbital interaction that has not been identified previously for any nitrosyl complexes.
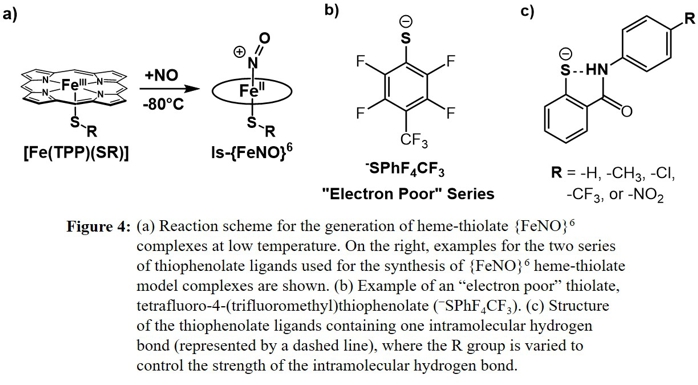
More recent work has finally proven this correlation between the thiolate donor strength and the Fe-NO and N-O bond strengths. For this purpose, we prepared a series of heme-thiolate {FeNO}6 model complexes
(all containing TPP2- as the coligand) in solution and characterized them by low-temperature UV-Vis, FT-IR and rRaman spectroscopy (see Figure 4). These complexes were prepared by reaction of the corresponding,
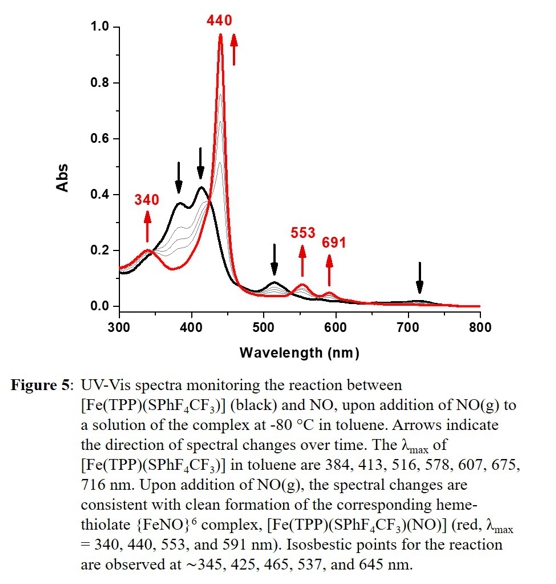
5C ferric heme-thiolate precursors with NO at -80 oC. Figure 5 shows an example for a typical reaction, followed by UV-Vis spectroscopy.
These studies have allowed us to analyze the effect of the proximal thiolate ligand on the Fe-N-O unit in a systematic and rigorous fashion. Through the use of thiophenolate ligands containing either (a) various electron
withdrawing substituents (i.e. the "electron poor" thiolate series) or (b) one intramolecular hydrogen bond to the thiolate sulfur (abbreviated -SPh-NHPh-pR, where R is a varied functional group
allowing for control of the hydrogen bond strength; see Figure 4b and c), it has been experimentally demonstrated that in heme-thiolate {FeNO}6 complexes:
(i) the thiolate donor strength directly modulates the Fe-NO and N-O bond strengths due to a thermodynamic σ-trans effect (interaction) that can be spectroscopically quantified by determination of the Fe-NO and N-O stretching frequencies;
(ii) either electron withdrawing groups or hydrogen bonds tune the thiolate donor strength in a similar manner;
(iii) hydrogen bonds provide additional protection for the thiolate ligand against S-nitrosylation, and likely, other detrimental side reactions (like protonation and reaction with O2); and
(iv) the collective strength of proximal hydrogen bonds to the thiolate ligand can in turn be gauged by determination of the Fe-NO and N-O stretching frequencies of the corresponding {FeNO}6 complexes, by
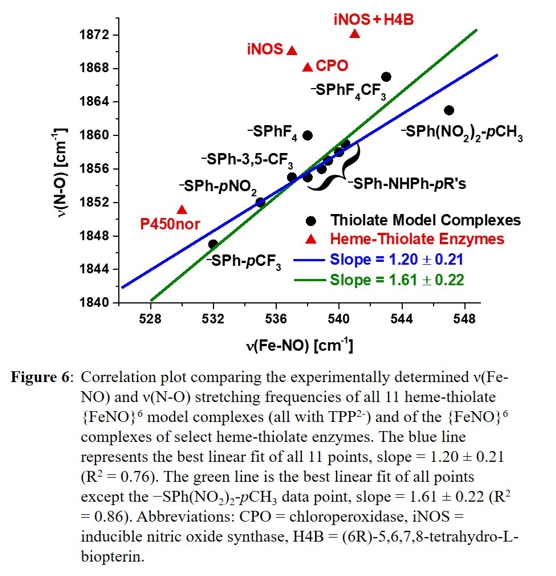
comparing those data to the vibrational correlation plot shown in Figure 6.
These experimental results are in excellent agreement with DFT calculations performed on Cyt. P450 heme-thiolate enzyme mimics, as well as the collective experimental results reported for different heme-thiolate enzymes (see above).
Now that these findings are firmly established by the experimental model complex studies described above, it can be concluded that the differences in Fe-NO and N-O stretching frequencies observed for different heme-thiolate {FeNO}6
enzyme complexes are in fact largely the result of a variation in the number and strength of hydrogen bonds to the cysteinate sulfur. Figure 6 shows the data points of 11 heme-thiolate {FeNO}6 model complexes and the two correlation lines,
compared to values reported for {FeNO}6 complexes in different heme-thiolate enzymes. The slopes obtained from the linear fits of the data points in Figure 6 (1.20 and 1.61) show a direct correlation of the Fe-NO and N-O stretching
frequencies, and hence, bond strengths, with the thiolate donor strength. For example, going from left to right in the plot, the thiolate donor strength decreases, leading to a simultaneous strengthening of the Fe-NO and N-O bonds. The
obtained slopes also suggest that the thiolate donor strength has a slightly greater effect on the N-O stretching frequency. However, by comparison of the relative (percent) change of the frequencies, the total variation observed for the Fe-NO
stretching frequency is in fact about three times larger (~3%) than that of the N-O stretching frequency (only ~1%). This suggests that the thiolate's donor strength has a greater impact on modulating the Fe-NO bond in heme-thiolate {FeNO}6
complexes compared to the N-O bond, which further supports the involvement of a thermodynamic thiolate σ-trans effect (interaction) to explain the experimental trend.
Through the use of DFT calculations, it was further proposed that the thiolate σ-trans effect identified in this way is equivalent to the underlying orbital interaction responsible for the "push effect" of the thiolate in Cyt. P450 monooxygenase
catalysis. The "push effect", first proposed by the Dawson and coworkers, is thought to accelerate heterolytic O-O bond cleavage in the protonated ferric-hydroperoxo intermediate (Compound 0) to give Compound I (a ferryl-oxo porphyrin radical cation
complex), which is the active oxidant responsible for C-H bond activation. Thus, the vibrational data of the {FeNO}6 complexes (which directly measure the thiolate donor strength) can also serve as a sensitive probe for the magnitude of
the "push effect" in Cyt. P450 monooxygenase enzymes. Because of this, NO is a suitable probe to evaluate the thermodynamic thiolate σ-trans effect (interaction) in new heme-thiolate enzymes and model complexes.
Reference:
V. K. K. Praneeth, E. Haupt, N. Lehnert
"Thiolate Coordination to Fe(II)-Porphyrin NO Centers"
J. Inorg. Biochem. 2005, 99, 940-948 (special issue: Heme-Diatomic Interactions, Part 2)
Erratum: J. Inorg. Biochem. 2005, 99, 1744
N. Lehnert, V. K. K. Praneeth, F. Paulat
"Electronic Structure of Fe(II)-Porphyrin Nitroxyl Complexes: Molecular Mechanism of fungal Nitric Oxide Reductase (P450nor)"
J. Comp. Chem. 2006, 27, 1338-1351 (special issue: Computational Bioinorganic Chemistry)
F. Paulat, N. Lehnert
"Electronic Structure of Ferric Heme Nitrosyl Complexes with Thiolate Coordination"
Inorg. Chem. 2007, 46, 1547-1549
L. E. Goodrich, F. Paulat, V. K. K. Praneeth, N. Lehnert
"Electronic Structure of Heme-Nitrosyls and Its Significance for Nitric Oxide Reactivity, Sensing,
Transport, and Toxicity in Biological Systems"
Inorg. Chem. 2010, 49, 6293-6316
(Inorganic Chemistry FORUM: The Coordination Chemistry of Nitric Oxide and its Significance for Metabolism,
Signaling and Toxicity)

N. Xu, L. E. Goodrich, N. Lehnert, D. R. Powell, G. B. Richter-Addo
"Preparation of the Elusive (por)Fe(NO)(O-ligand) Complex by Diffusion of Nitric Oxide into a Crystal of the Precursor"
Angew. Chem. Int. Ed. 2013, 52, 3896-3900
A. Rana, S. Amanullah, P. K. Das, A. B. McQuarters, N. Lehnert, A. Dey
"A Formally Ferric Heme Carbon Monoxide Adduct"
J. Am. Chem. Soc. 2019, 141, 5073-5077
A. B. McQuarters, E. J. Blasei, E. E. Alp, J. Zhao. M. Hu, C. Krebs, N. Lehnert
"Synthetic Model Complex of the Key Intermediate in Cytochrome P450 Nitric Oxide Reductase (P450nor)"
Inorg. Chem. 2019, 58, 1398-1413
A. P. Hunt, N. Lehnert
"The Thiolate Trans Effect in Heme {FeNO}6 Complexes and Beyond: Insight into the Nature of the Push Effect"
Inorg. Chem. 2019, 58, 11317-11332
(selected for Journal cover: Issue 17, Sept 02, 2019)

J. Tejero, J. Santolini, A. P. Hunt, N. Lehnert, D. Stuehr
"Mechanism and regulation of ferrous heme-nitric oxide (NO) oxidation in NO synthases"
J. Biol. Chem. 2019, 294, 7904-7916
M. R. Dent, M. W. Milbauer, A. P. Hunt, M. M. Aristov, I. Guzei, N. Lehnert, J. N. Burstyn
"Electron Paramagnetic Resonance Spectroscopy as a Probe of Hydrogen Bonding in Heme-Thiolate Proteins"
Inorg. Chem. 2019, 58, 16011-16027
A. P. Hunt, S. Samanta, M. R. Dent, M. W. Milbauer, J. N. Burstyn, N. Lehnert
"Model Complexes Elucidate The Role of the Proximal Hydrogen-Bonding Network in Cytochrome P450s"
Inorg. Chem. 2020, 59, 8034-8043
Literature:
[1] 'Nitric Oxide Research from Chemistry to Biology: EPR Spectroscopy of Nitrosylated Compounds';
Henry, Y. A.; Guissani, A.; Ducastel, B., eds., R. G. Landes Company, Austin, 1997.
[2] Praneeth, V. K. K.; Neese, F.; Lehnert, N. Inorg. Chem. 2005, 44, 2570-2572
[3] Patchkovskii, S.; Ziegler, T. Inorg. Chem. 2000, 39, 5354-5364.
[4] O'Keeffe, D. H.; Ebel, R. E.; Peterson, J. A. J. Biol. Chem. 1978, 253, 3509-3516.
[5] Shiro, Y.; Fujii, M.; Isogai, Y.; Adachi, S.-I.; Iizuka, T.; Obayashi, E.; Makino, R.; Nakahara, K.; Shoun, H.
Biochemistry 1995, 34, 9052-9058.
[6] Cooper, C. E. Biochim. Biophys. Acta 1999, 1411, 290-309.
[7] Xu, N.; Powell, D. R.; Cheng, L.; Richter-Addo, G. B. Chem. Commun. 2006, 2030-2032.
|
