Welcome the new group member
Welcome Zongwei, Callie and Pouya !

Welcome new group members!
Continue ReadingDepartment of Chemistry
Introduction to research in the Geva Group
We are a theoretical and computational chemistry group in the University of Michigan, Ann Arbor
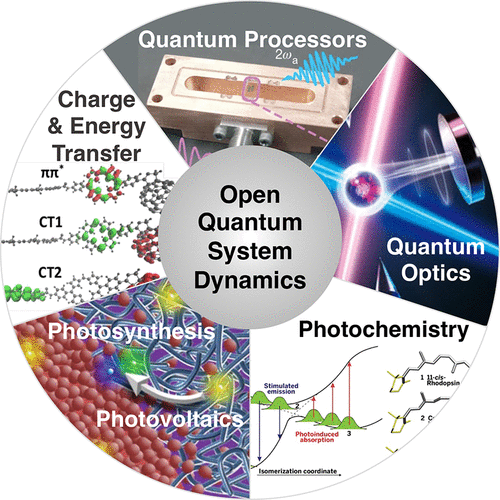
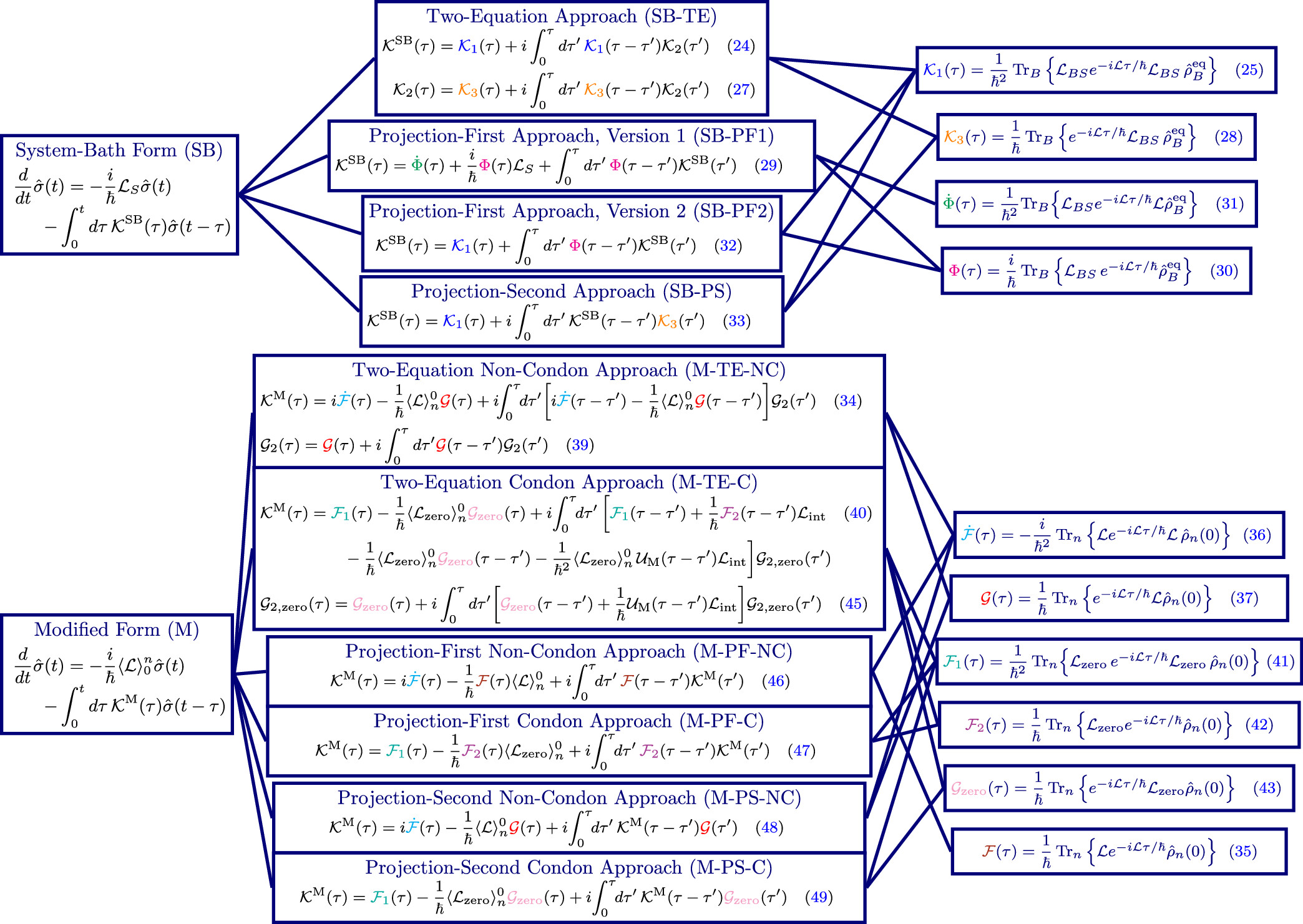
Research in the Geva group revolves around the development and application of models and computational techniques for simulating decoherence and the dynamics of chemical processes in complex molecular systems, and their time-resolved nonlinear spectroscopic signature. To this end, a variety of theoretical approaches are explored, advanced and used, including quantum master equations, semiclassical approximations, quantum rate theory and optical response theory.
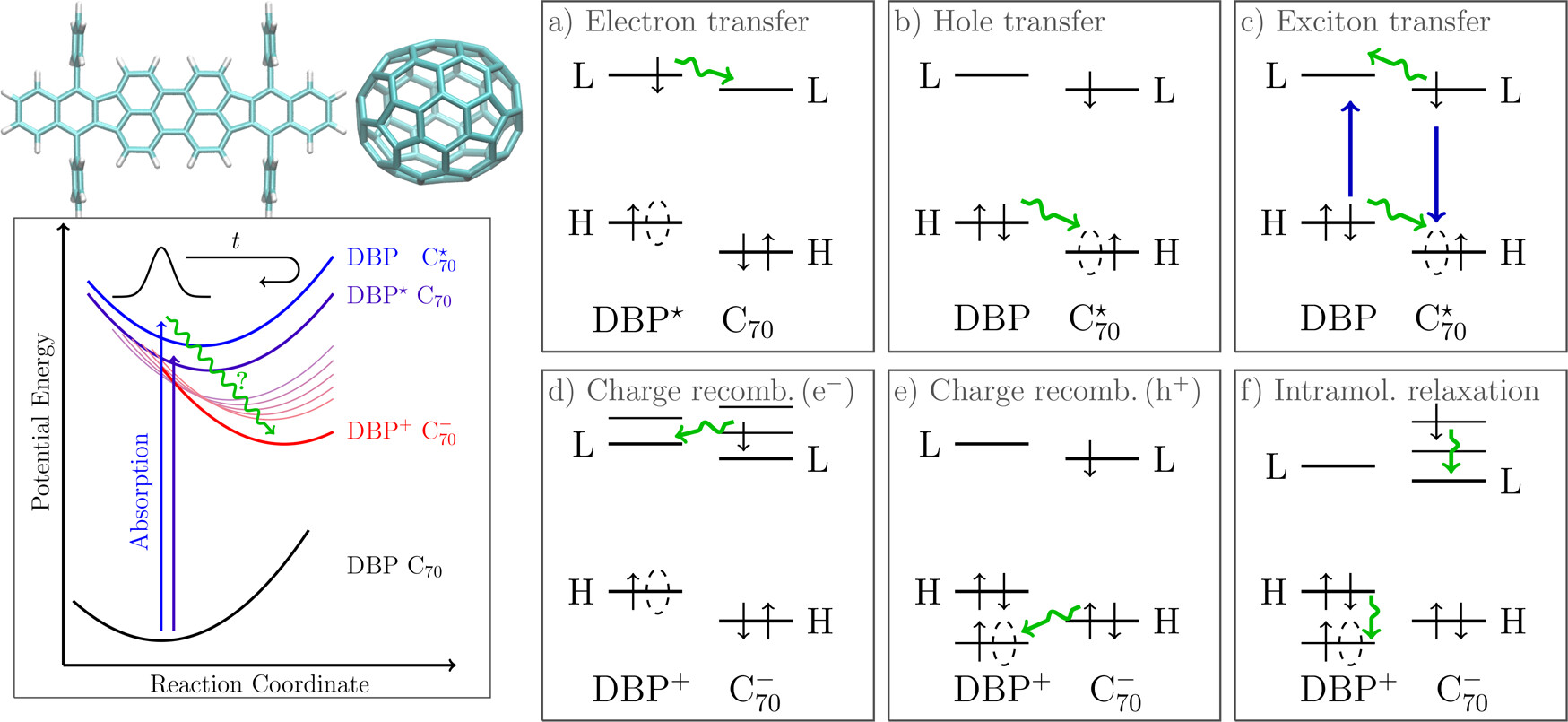
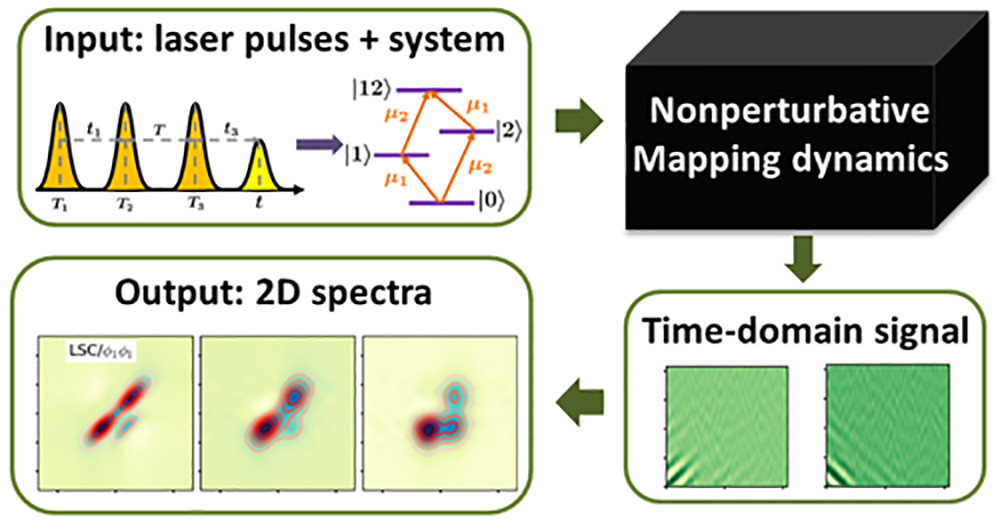
Current and ongoing research projects include the development of computational tools based on the generalized quantum master equation and its various approximate versions for simulating electronic energy and charge transfer, and their application to the emerging fields of quantum science and technology, polaritonic chemistry and organic photovoltaics.
Welcome Zongwei, Callie and Pouya !
Welcome new group members!
Continue Reading