Amino Acid Metabolism
(Nitrogen Metabolism) Dec
9-11 2008 Dr.
Robert Lyons
See: MCB500 for supplementary (non-required) course materials.
Medical relevance of amino
acid metabolism pathways:
What is nitrogen balance, and what affects it?
Role of vitamins: pyridoxamine (VitB6), folic acid
Understanding a critical function of the liver: nitrogen metabolism
Which amino acids are essential?
Inborn errors of metabolism: amino acid breakdown, urea cycle
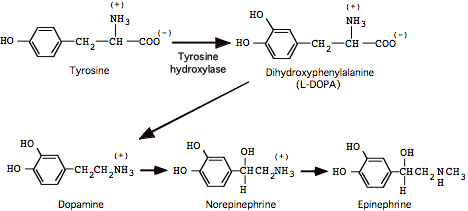
Pharmacologic manipulation of neurotransmitters (e.g. Parkinson's Syndrome)
I. Protein degradation/Nitrogen balance
A. Cells constantly turn over proteins
It's a normal process, balanced by protein intake.
Proteins can be degraded if they are:
damaged by free radicals
oxidative damage
misfolded
no longer needed.
B. "Nitrogen Balance" expresses the balance
between anabolism and catabolism
1. Measured by
assessing dietary N intake vs urinary N output (as urea)
2. "Positive"
nitrogen balance (net storage of nitrogenous compounds):
childhood growth
pregnancy
muscle building
healing
3. "Negative"
nitrogen balance (net breakdown of stored nitrogenous compounds):
illness
uterine resorption
starvation
amino acid deficiency
wounding
4. In negative nitrogen balance, the liver may be taxed in
handling excess nitrogenous waste. We will revisit this when we discuss
pathologies of the nitrogen disposal pathways.
II. AA catabolism - Separate the amino moiety, carbon skeleton:
A. Dealing with the amine: convert to urea or re-utilize:
1. transaminated to a-ketoglutarate forming an a-keto acid and glutamate.
Simplified version:
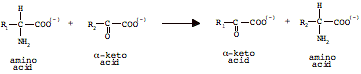
Complicated version, step 1: Transfer amine to
pyridoxal phosphate (PLP)
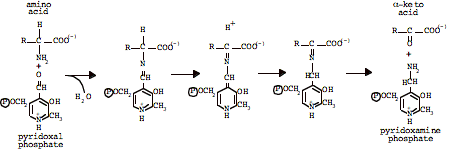
Complicated
version, step 2: Transfer amine to acceptor a-keto
acid:
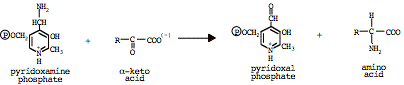
In peripheral tissues, catabolism
of amino acids tends to form glutamate (i.e. a-ketoglutarate
is the preferred N-acceptor)
2. The amino group on glutamate can be transferred back to another keto-acid if needed by reversing the above reactions. A specific enzyme in liver mitochondrial matrix (Glutamate-aspartate aminotransferase) catalyzes exchange of amine groups between glutamate and aspartate. (Reaction depicted further down)
3. To discard, the amino group in glutamate is transported to the liver via glutamine:
a. Release amino group as ammonia (enzyme: glutamate dehydrogenase, oxidative deamination):
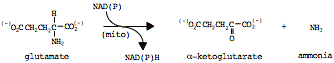
b. Incorporate the ammonia on a separate glutamate to form glutamine (enzyme: glutamine synthetase):
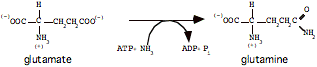
c. Glutamine passes through cell membranes via a variety of transports mechanisms, and into the bloodstream, to be taken up by other tissues, most notably (for the purposes of the current discussion) the liver.
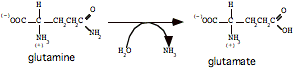
d. In the liver, the enzyme glutaminase releases ammonia by hydrolysis of glutamine, leaving glutamate (also occurs in kidney and intestine).
The glutamate can go on to form aspartate, via glutamate-aspartate aminotransferase:
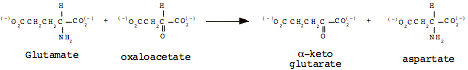
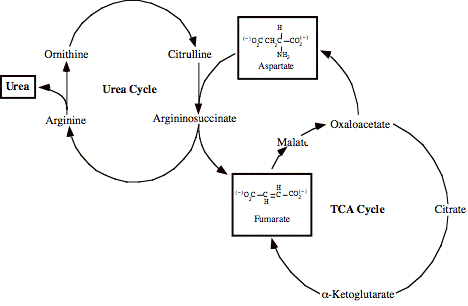
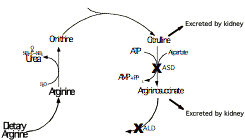
e. The urea cycle (only in liver) makes amine moieties into urea for excretion
Executive Summary: Amine groups are assembled onto ornithine (which essentially acts as a 'handle'), and they are ultimately cleaved off as Urea (releasing the ornithine again). This occurs in the liver, partly in the mitochondrial matrix and partly in the cytoplasm.
Key amino compounds entering the Urea Cycle:
Aspartate (typically from transamination of oxaloacetate; see IIA2 and IIA3d, above)
Ammonia, which may come from many sources, especially hydrolysis of glutamine (see IIA3a, above) and oxidative deamination of glutamate (see IIA3d, above). See also breakdown of purines, in a later lecture.

[this page intentionally left blank]
Step 1: Formation of cabamoyl phosphate.
Catalyzed by Carbamoyl Phosphate Synthetase I (CPS I), in liver mitochondria
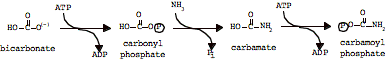
Step 2:
Formation of Citrulline
Catalyzed by Ornithine Trans-Carbamoylase (OTC), in liver mitochondria
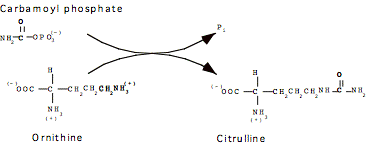
Step 3:
Formation of Argininosuccinate
Catalyzed by Argininosuccinate Synthetase (AS), in liver cytoplasm
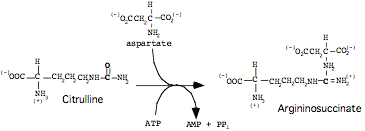
Step 4:
Cleavage to form Arginine
Catalyzed by Argininosuccinase, a.k.a. Argininosuccinate Lyase (AL), liver cytoplasm
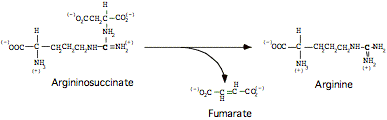
Step 5:
Cleavage to release Urea
Catalyzed by Arginase (no abbreviation); liver cytoplasm
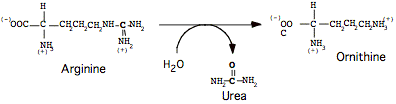
f. Regulation and energetics of the urea cycle
(i) Rule of thumb: Any reaction that creates a new C-N bond costs one ATP.
- one ATP spent to put the amine on carbamoyl phosphate
- one ATP spent to put the phosphate on CP, which drives the next reaction (formation of citrulline; OTC)
- one ATP (*two* Hi-E bonds) used to make argininosuccinate.
(ii) The urea cycle costs energy BUT it produces energy as well:
- Fumarate is oxidized in the Krebs cycle to Oxaloacetate, yielding one NADH (which can be used to make three ATP).
- The glutamate dehydrogenase reaction also yields NADH (also equivalent to three ATP).
(iii) Energy-requiring steps are often regulated:
- The CPS step is allosterically stimulated by N-acetyl glutamate (formed by the enzyme N-Acetyl Gluamate Synthetase [NAGS], in mitochondrial matrix).
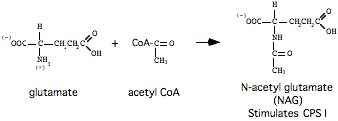
- The CPS step is also postulated to respond to the levels of ammonia in the mitochondrion.
- The other enzymatic steps are apparently governed only by their substrate concentrations.
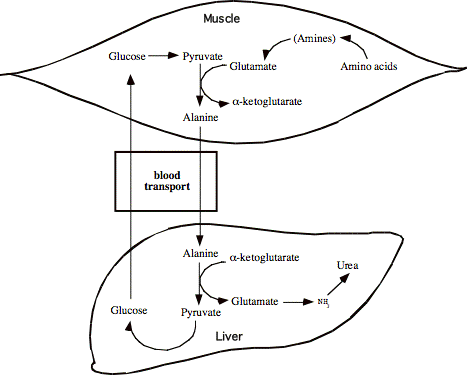
g. Special role for alanine in energy metabolism in muscle: the glucose-alanine cycle.
Muscles frequently utilize amino acids as energy sources, They are consequently particularly active for production of glutamine.
Under heavy energy demands, muscles convert to anaerobic energy production via simple glycolysis, producing excess pyruvate and lactate.
The excess pyruvate and ammonia can be converted to alanine and sent to the liver. There the amino groups are converted to urea and the pyruvate is used in gluconeogenesis to form glucose, which goes back to peripherals via blood.
h. Other organs involved in nitrogen metabolism:
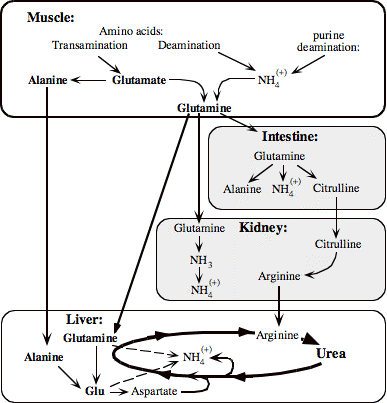
i. Kidneys can break down glutamine:
- More acidic conditions increase that breakdown, less acid decreases it.
- The resulting ammonia is protonated to form ammonium ion, and excreted in the urine.
- This is thought to be a way to compensate for acidosis
ii. Kidneys and intestines jointly produce arginine
- Intestinal CPS 1 and OTC form citrulline, exported in the
blood
- Kidneys take up citrulline, and forms arginine (using Argininosuccinate
synthetase), which is again exported to the blood
- This mechanism is primarily used to synthesize Arg for
purposes OTHER than ureagenesis, e.g. protein synthesis (see also other
products of Arg, later).
- Massive resection of the small intestine can cause patients to become Arginine deficient
Sidebar: Why is ammonia toxic?
(speculation only!)
Glutamate level is disturbed; and since it
is a neurotransmitter, its levels may be critical to proper neural function.
Glutamate is recycled from post-synaptic
neuron to pre-synaptic neuron as glutamine (via glutamine synthetase), and that
step is probably disturbed by high ammonia levels.
Glutamate is also the precursor another
neurotransmitter, gamma aminobutyric acid (GABA), which thus may be affected by
hyperammonemia.
Alterations in glutamate levels may
influence energetics. In addition, removing ammonia uses ATP (glutamine synthetase), also with
potentially detrimental effects on energetics.
B. Inborn Defects of Nitrogen Metabolism (Urea Cycle)
1. Generalized features of urea cycle defects:
a. Loss of an enzyme causes substrate to build up
The pathway backs up all the way to ammonia, which is toxic
b. Complete absence of any of these enzymes causes neonatal death
c. Classic presentation:
Infant displaying irritability, hypotonia, lethargy, vomiting, ataxia, delayed growth
Older child or adult displays similar symptoms after precipitating event (e.g. feast). If untreated, progresses to spasticity, mental retardation, coma, death.
2. Diagnosis:
a. All of the deficiencies may present with hyperammonemia
b. Determine which enzyme by substrate concentrations:
CPSD: only hyperammonemia; diagnose by elimination
OTCD: hyperammonemia with orotate in blood, urine
ASD: elevated citrulline in blood, urine
ALD: elevated argininosuccinate in blood, urine
AD: elevated arginine in blood, urine
3. Treatment:
a. Dialysis to reduce the blood ammonia levels
b. Intravenous sodium benzoate and phenylacetate to provide for nitrogen disposal
(both compounds bind amino acids and are then excreted.)
c. Supplementation of arginine
d. Minimize gut production of ammonia - lactulose or antibiotics
e. Low protein diet (long-term solution)
C.
Amino acid carbon backbone - degraded for metabolic fuel
1. Glucogenic versus ketogenic amino acids
2. Sometimes mere transamination can produce easily-degraded compounds:
a) aspartate forms oxaloacetate
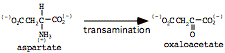
(can also degrade via Urea cycle)
b) glutamate forms alpha-ketoglutarate
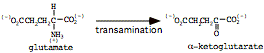
(can also degrade via oxidative deamination)
c) alanine forms pyruvate
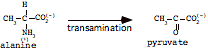
3. glutamine and asparagine deaminated to glutamate and aspartate (as above).
a) glutaminase reaction: glutamine + H2O --> glutamate + NH4+

b) asparaginase reaction: similar to glutaminase, produces aspartate.
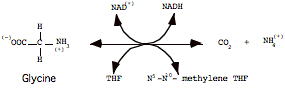
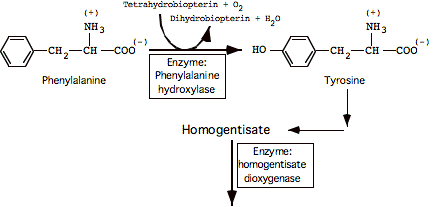
4. Other amino acids: see following figures:
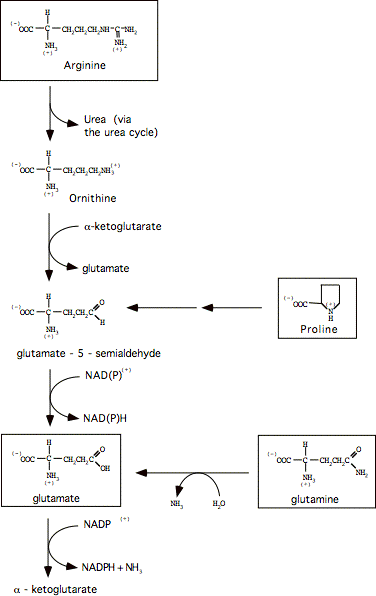
Serine hydroxymethyl transferase:

Glycine Synthase (a.k.a. Glycine Cleavage System):
Serine dehydratase:
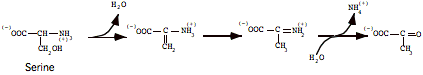

Degradation of Phenylalanine and Tyrosine:
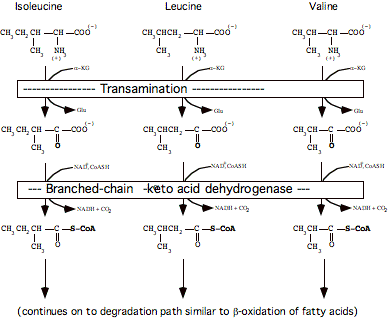
Branched-chain amino acids:
D. Precursors
for other important biomolecules - bioactive amines
Tyrosine:
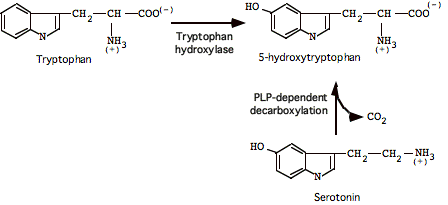
Tryptophan:
Glutamate:

Histidine:

E. A few other important products derived from amino
acids:
Arginine is converted to NO, Agmatine, creatine and creatinine
Glutamine, cysteine and glycine are used to make glutathione
Glycine is used in biosynthesis of heme
Glutamine, glycine and aspartate contribute to purine nucleotide synthesis
Aspartate is used in pyrimidine nucleotide biosynthesis
III. Amino acid biosynthesis
Essential vs. non-essential amino acids:
A. Non-essential - we can synthesize adequate amounts of:
Alanine, Glycine, Serine, Glutamate, Aspartate, Glutamine, Asparagine, Proline, (Cys, Tyr - usually considered non-essential, but it's semantics - depends on sufficient Met, Phe)
B. Essential: all the rest - Arginine (conditionally), Phenylalanine (enough to make Tyrosine, too), Methionine (enough to make Cysteine as well), Histidine, Isoleucine, Leucine, Lysine, Threonine, Valine, Tryptophan.
For each of the non-essential amino acids listed above, review the degradation pathways already discussed to identify the probable biosynthesis route (skip proline - that wasn't really covered).